CH637Computational Organic Chemistry(
CRN 27694 )Exercises |  | |
 | ||
General policies
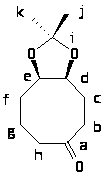
Exercise 1. Due 1/14. 10 points 1. Create your "Homework" directory on wngr343d. Place this file in your Homework directory: kpgtest.gjf. 2. Copy this file to another filename, and convert it from a Gaussian Z-matrix file to a Mopac cartesian input file using Maestro. Use the .dat extension. 3. Work Experiment 1 in HSH. Instead of a semiempirical calculation, perform the exercise using 3-21G ab initio calculations. Next, build 1,3,5-cyclohexatriene by going into the Builder and fix the C-C bonds at alternating lengths of 1.50 and 1.30 angstroms ("Geometry", then "Constrain Distance"). Rerun the optimization, using the constraints by checking the appropriate box in "Setup". Compare the energies and consider them in light of the "aromatic stabilization energy" of 33 kcal/mol for benzene. Be sure to convert the energy difference from Hartrees to kcal/mol (1 Hartree = 627.5 kcal/mol). Generate electrostatic potential maps for your two molecules and map them on the same scale. What differences do you see? 4. Work Experiment 43 in HSH, as well as the Optional section. Build norcamphor (2-oxo-bicyclo-[2.2.1]-heptane) and optimize at the AM1 semiempirical level. Once the structure is optimized, perform a single point energy calculation at the 3-21G ab initio level. Generate the LUMO by selecting "Setup"; "Surfaces" and adding LUMO with the property LUMO to the list. Display this on the molecule. (Type "1" to generate a solid surface if you are accessing remotely.) The LUMO shows where a nucleophile is likely to attack. Is there a difference in the two faces of the ketone in terms of the shape of the LUMO? Experimentally, reduction with NaBH4 leads to 86% of the endo isomer (from exo attack of hydride). Exercise 2. Due 1/21. 10 points A. Find the conformational minima for the following molecule using the MMFF force field (or the closest available choice in your software):
1. How many unique structures are there within 3.0 kcal/mol of the global minimum? 2. Calculate (using the Boltzmann distribution) the proportions of each of these minima present at 25ºC (298K). 3. NMR measurements reveal nOe between the two hydrogens at the ring junction (d and e) and methyl group k. For each of your minima, list the closest distance between hydrogens at k and both d and e. What do your calculations reveal about the nOe phenomenon in this case? B. Find the rotation barrier for the aryl-aryl bond in the following compound: 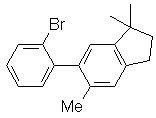 Build the molecule and minimize it (MM/MMFF). In Spartan, you can go to "Build", then "Define Profile". Select "Dihedral", then select the four atoms that define the dihedral angle. You will want to drive the dihedral from appr. +90º to -90º or from +90º to +270º (depending on the direction of rotation). Save the molecule, then set up calculations for an Energy Profile, using MM/MMFF as the method/force field. Other programs will have similar ways to run the experiment; check documentation. 1. Does it matter which direction the rotation goes? Explain. 2. The experimental activation barrier is 15.8 ± 0.4 kcal/mol. Examine your structures closely and explain whether any discrepancy lies with the experimental measurement or with the way the computational model was constructed. Exercise 3. Due 1/28. 10 points 1. Work Experiment 2 in HSH. Also, describe how the Huckel approach would have to be modified to address the question of whether thiophene is aromatic. 2. Work Experiment 7 in HSH. Use HF/6-31G* for final geometry optimization/energies. Answer questions in the "Optional" section. Exercise 4. Due 2/4. 20 points 1. Experiment 6 in HSH. 2. Experiment 47 in HSH. Include the Optional section on tropene. 3. Huckel calculations; located on a separate page; turn in hard copies of your work by 5 p.m Friday. Exercise 5. Due 2/11. 10 points 1. Work Experiment 5 in HSH. Students with family names beginning with A-L should use AM1, while those whose family names begin with M-Z should use PM3. We will compare the outcomes during class discussion. 2. Work Experiment 9 in HSH. Use the 6-31G* basis set in place of 3-21G. Use the default "Solvation Energy" option in Calculation Setup where called for. Those who are curious may compare the current solvation model to the older AM1-SM2 model by placing the keyword SOLVENT=SM2 in the "Options" box.3. Work Experiment 19 in HSH. Exercise 6. Due 2/25. 20 points 1. Work Experiment 17 in HSH. Perform the Optional portion #1, and compare geometries and energies at the AM1, 3-21G and 6-31G** level. Explain how the choice of method affects the results, both qualitatively and quantitatively. 2. Work Experiment 22 in HSH. Select a method (AM1, 3-21G or 6-31G**) based on your observations from Experiment 17, and explain the basis for your selection. Include both Optional portions. Exercise 7. Due 3/4. 10 points Caclulate the energy of ionization for tert-butyl chloride and benzyl choride at the AM1 level by computing heats of formation of the reactants, the carbocations, and chloride ion. For each optimized species, calculate the CI stabilization. In Spartan, use the default 6-level CI calculation by inserting the CI keyword and performing a single point calculation. Does the use of CI have a significant impact on your prediction for the relative reactivities of these two reactants for SN1 substitution? Exercise 8. Due 3/11. 10 points 1. Work Experiment 71 in HSH. Use B3LYP/6-31G** to establish final structures and energies.
Back to CH 637 Home Page |