Important parameters
The following is a list of some of the most frequently used
parameters.
Namelist allatoms
See under namelist atom: dx, jri.
Namelist atom
econfig
electronic config: program will pick a reasonable configuration;
core/valence states separated by ‘|’; scalar core states have a ‘s’ appended
Examples:
B: econfig='1s2 | 2s2 2p2'
B 1s-corehole: econfig='1s1 | 2s2 2p2'
Co: econfig = '[Ar] | 3d7 4s2'
Co, scalar 3p: econfig = '[Ne] 3s2 3p6s | 3d7 4s2'
To specify (core) occupations of relativistic levels, give j value:
econfig='1s2 2s2 2p(1/2)1 2p(3/2)4 | 3s2 3p2'
By default core holes are put in the highest j level.
element
atom name
Example: element=’Ni’
id
atom identifier used in atomic positions
rmt
muffin-tin radius
Namelist comp
gmax
max. G-vector (in a.u.^{-1}) for density/potential
kmax
max. k for wave functions
jspins
number of spins; =2 for spin-polarized
Namelist conv
itmax
max. number of iterations for total run
itmax_scf
max. number of iterations for each atomic step
Namelist exco
Exchange-correlation: either lda or gga. Default is lda=’pz’.
lda
xa, wign, hl, bh, mjw, vwn, pz
gga
l91, pw91, pbe, rpbe, wc
Namelist geo
l_geo
do geometry opimization
maxstep
max number of optimization steps
Namelist kpt
div1
division along
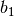
reciprocal lattice vector
div2
division along
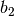
reciprocal lattice vector
div3
division along
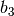
reciprocal lattice vector
kshift
shift mesh away from zone center if symmetry allowed
Namelist out
bsplot
band structure calculations (then need file bskpts and also
the namelist &band
dosplot
calculate dos and generate dos plot files