Main input file: inp¶
The set-up and order of the input file is as follows; information about the namelists can be found here: All namelists.
The first (non-blank) line is the title not starting a ‘&’. (optional)
namelist &input (optional)
lattice information input by: (required)
- &lattice (see fcc Cu example below) or
- explicitly, with the lattice vectors given in ‘scaled’ cartesian coordinates:
For a bulk system the explicit specification of a lattice is as follows:
1 2 3 4 5 6 | a1(x) a1(y) a1(z) ! components of a1
a2(x) a2(y) a2(z) ! components of a2
a3(x) a3(y) a3(z) ! components of a3
a0 ! overall lattice constant (in atomic units)
scale(1:3) ! scale factors for each direction if scale < 0,
then take square root, i.e., -3 = sqrt(3).
|
In line 4 the volume of the unit cell can be specified instead:
vol= vol ! volume of cell
To give the lattice constant or volume in Angstroms, append an “A”, or set angstrom=t in &input. Note that fractions, rather than their decimal equivalents, can be given; for example 1/3 rather than 0.33333... is fine.
For a film calculation, line 3 changes to:
a3(x) a3(y) a3(z) dvac ! components of a3 and dvac for film
As an example, a hexagonal lattice could be input as:
1/2 -1/2 0 ! a1
1/2 1/2 0 ! a2
0.0 0.0 1 ! a3
6.2 ! lattice constant
0.0 -3.0 1.62 ! scale(1) is 1 by default
which corresponds to the lattice vectors (in Cartesian coordinates):
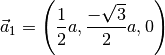
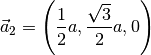
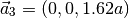
Atomic positions:
- number of atoms in input (required)
- For each atom: id pos1 pos2 pos2 (required) where id is an identifer (and default atomic number) and the atomic positions either in lattice or scaled cartesian coordinates (see &input ) Zero or more namelist(s) &origin can be inserted; all positions after that will have origin(:) added to each component
- &shift shift(1) ... / (optional)Added shift to all atomic positions
- &factor factor(1) ... / (optional) factor(s) to divide each component by
- &gen or &sym (optional) symmetry given either by the generators of the group ( &gen ) or by all operations ( &sym )Next comes the information about the atoms . The id atomic identifer is used to relate the information to each atom; the default is to use id as the atomic number.
- &allatoms (optional) information common to all atoms
- &atom (optional) information for particular atomsThe rest of the input may be written to para or to inp.
- For any other namelist see all parameters.; the order is generally not important. (optional)
- &end (optional) stop reading the input; everything after this point ignored.
As an example of how easy/compact an input file can be, the following is all that is needed to run fcc Cu:
Cu (fcc)
&lattice bravais='cF' a0=6.50 /
1 ! number of atoms
1 0 0 0
&atom id=1 element='Cu' /
Be aware that the defaults for k-points and iterations are probably not what you want, although the program will pick values that should give you a reasonable result. More commonly, one will give more information such as in this input:
GeMnN_2 (oP16 structure) (Antiferromagnetic)
&input cartesian=t checkinp=f /
1 0 0
0 1 0
0 0 1
10.367 ! lattice constant
1.0 1.2167 0.95625
16 ! number of atoms
1 0.070 1/8 0
1 -0.070 -1/8 1/2
1 0.570 3/8 0
1 -0.570 -3/8 1/2
2 0.083 -3/8 0
12 -0.083 3/8 1/2
12 0.583 -1/8 0
2 -0.583 1/8 1/2
3 0.071 1/8 0.366
3 -0.071 -1/8 0.866
3 0.571 3/8 0.366
3 -0.571 -3/8 0.866
3 0.083 -3/8 3/8
3 -0.083 3/8 7/8
3 0.583 -1/8 3/8
3 -0.583 1/8 7/8
&allatoms jri=361 lnonsph=4 dx=0.028 /
&atom id=1 element='Ge' rmt=2.10 / ! econfig='[Ar] 3d10 | 4s2 4p1' /
&atom id=2 element='Mn' rmt=2.20 /
&atom id=12 element='Mn' rmt=2.20 /
&atom id=3 element='N' rmt=1.40 /
&comp gmax=12.0
kmax=3.75
jspins=2
/
&swsp 2=4 12=-4 /
&kpt tkb=0.001 div1=8 kshift=t /
&mix alpha=0.3 /
&conv itmax_scf=50 /
&geo l_geo=t maxstep=6 /
&out dosplot=t /
!&exco lda='pz' /
&exco gga='pbe' /
This sets up an antiferromagnetic system. Note that atoms with id=2 and 12 are the same; they are given different identifiers to force the symmetry program to treat them differently. The &swsp namelist starts the spin-polarized calculation with moments on atom with id=2 and 12. Note the use of comments in the input. At the end of the run, the density of states will be generated. Different choices for exchange-correlation are listed, but commented out, so the default will be used.