Answers to Problem Set 5
4.7.
a. Rate = (k1k2/k-1){[S][R3N]/[R3NH+]}
Apply SSA to HCCBr, and equilibrium condition to the vinyl anion.
b. You need to define rate.
In terms of the individual products:
d[ROH]/dt = {k1k2[S][H2O]}/{k-1[-OPNB] + k2[H2O] + k3}
d[Alkene]/dt = {k1k3[S]}/{k-1[-OPNB] + k2[H2O] + k3}
Note that if k-1[-OPNB] is presumed to be small, these add together and simplify to:
-d[ROPNB]/dt = k1[S].
(S is substrate in this example.)
c. If the sigma complex is a steady-state intermediate, then k1 is slow.
d[P]/dt = {k1k2[S][Br2]}/{k-1[Br-] + k2}
Simplification requires estimating the relative magnitude of k-1[Br-] and k2; the rapid equilibrium of the final step indicates that [Br-] is always small and therefore k2 dominates. Thus, the simplified rate expression is:
d[P]/dt = k1[S][Br2]
d. The restriction to make no assumption about relative magnitude of rate constants requires that you analyze both the possibility that k1 is slow and that k2 is slow.
k1 slow: d[P]/dt = {k1k2[S][Cr]}/{k-1 + k2[Cr]} (by application of the SSA to the enol) If k-1 is small, this reduces to k1[S].
k2 slow: d[P]/dt = {(k1k2/k-1)[S][Cr]} Note that in the above expression, assuming k2 is small (our condition here), we get the same expression.
(Again, S is the keto substrate.)
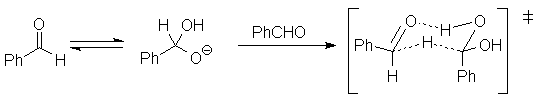
4.11. The minimal interpretation of each piece of data is:
A model for the transition state consistent with this data is:
5.6. The data provided can be minimally interpreted as follows:
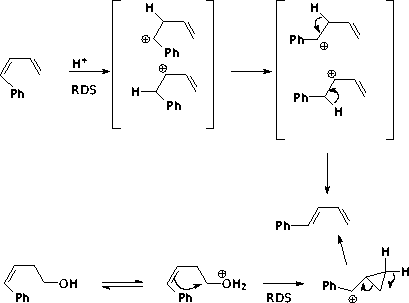
The alcohol must go through the cyclopropylcarbinyl species in order to avoid a primary carbocation. An alternative is to eliminate to the Z diene, then isomerize as above, since the Z diene isomerizes faster than the alcohol.
5.8. This system is controlled exclusively by the timing of nucleophilic attack relative to formation of tight ion pairs, solvent-separated ion pairs, and free ions. Species 2 arises from neighboring group participation by the double bond; attack of the nucleophile on the cyclopropane (at the original C-OTs site) gives 1 with retention. Attack of the tight ion pair (prior to interaction with the double bond) gives inversion. From the rate data, we can surmise that the RDS is always generation of the TIP.
6.13. Addition of Br+ can occur from either face; anti addition of bromide leads to the two expected trans products. If Br+ addition occurs anti to the acetate, intramolecular neighboring group participation leads to formation of two unexpected cis dibromides via a bicyclo-[4.3.0] system.
Back to Homework page
Back to CH 630 Home page
Last updated: 10/09/03