Answers to Problem
Set 4
4.2
- Phosphorus-for-Cl
substitution (SN1 or SN2!),
followed by attack of chloride on methyl ( SN2 ).
- Form the activated OBs ester; on ionization migrate the bond
that is aligned and behind.
- Protonate the alkyne; LP from Cl attacks to form a cyclic
intermediate. Note: trifluoroacetate is not nucleophilic enough to
displace chloride unless it is in this bridging arrangement!
- First case: Diazotization; ionization accompanied by alkyl
migration to
form resonance-stabilized oxocarbocation. Second case: The
locked conformation (tBu) prevents
alkyl migration as above; now hydride is anti-periplanar and migrates.
- A norbornyl-like rearrangement gives an oxocarbocation that
on addition of water gives product as the hemiacetal form.
- Neighboring group participation by N gives a symmetric
intermediate. Note that exo and endo isomers are diastereomeric, not
enantiomeric!
- Neighboring group participation by the amide C=O oxygen forms
a cyclic intermediate.
4.3.
- Fluorinated sulfonate a
better LG.
- CH3 more electron rich; stabilizes
carbocation.
- Both would require substantial deformation of the strained
norbornyl ring system; the 7-tosylate has a sigma bond aligned in the
back that sets up a rearrangement.
- Moving the tBu group provides relief
of steric strain.
- The double bond cannot conjugate, and the sp2
center makes rehybridization more difficult.
- This is an SN2 reaction; which the
sulfonyl makes the attached carbon more electrophilic, it also adds
steric bulk. The aactual rate ratio is 38:2.
- Cyclopropylcarbinyl; the first example is meopentyl and
undergoes a slightly slower but concommitant rearrangement to a
tertiary cation.
- Presence of the alkene provides neighboring group
participation & formation of the 2-norbornyl ion.
- Neighboring group participation; 5-member ring >
4-member ring.
- Neighboring group participation by the ortho
acetate.
4.13. a. Participation of the aromatic electrons is more
costly than of the C=C pi electrons.
c. Two bond migrations
compete in each case. One leads
to a cyclopropylcarbinyl
cation, differing for the two systems in being perpendicular or
parallel at the transition
state. This predominates for E because of the parallel orientation.
Look at models for the two compounds (different rotamers) and
the two ions:
|
|
|
| From anti isomer: Migration of C-1
to form the C-3 cation:
415c_1a.mol
|
From the anti isomer: Migration of C-3 to form the C-2 cation:
415c_2a.mol
|
|
|
|
| From the syn isomer: migration of
C-1 to form the C-3 cation:
415c_1s.mol
|
From the syn isomer: migration of C-3 to form the C-2 cation:
415c_2s.mol
|
|
|
|
| The product from the anti isomer:
1_310.mol
|
The product from the syn isomer:
2_310.mol
|
F would lead to a
perpendicular
cyclopropylcarbinyl cation, so it does the other
migration, leading to a nonclassical cation delocalized into the
bridgehead-bridgehead
cyclopropane bond.
B. Additional problems.
1.Polar solvent in red; nonpolar in black.
a. Ionic reactants, neutral TS/product.
Stabilize reactants, increase activation barrier and slow rate in polar
solvents.
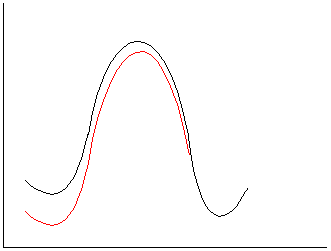
b.
Polar reactants, TS and products. Little change, either a
slight increase or slight decrease in rate could be rationalized.
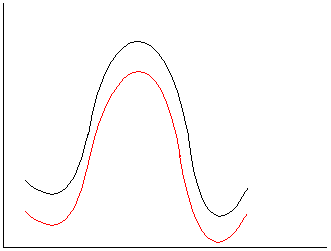
c.
Neutral reactants; polar TS/products. The stabilization of
developing charge will assist the formation of the transition state and
accelerate the reaction.
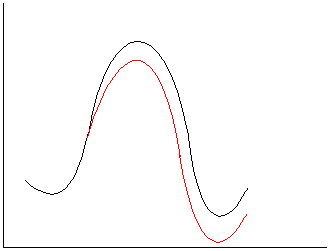
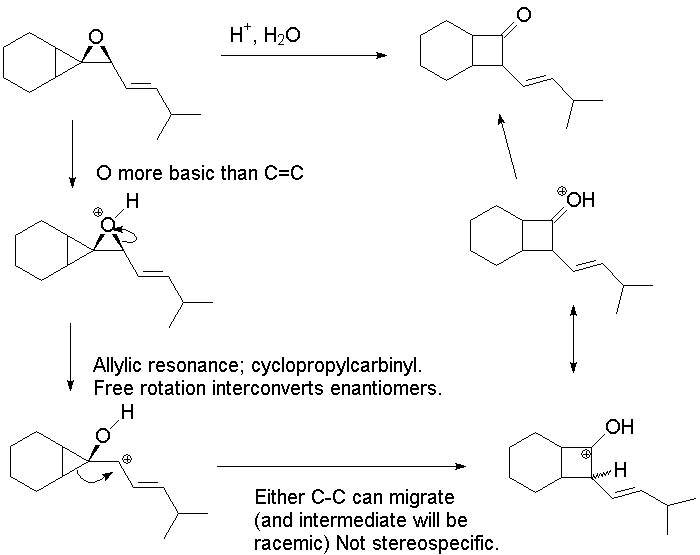
2.
Back to Homework
page
Back to CH630
Home page
Last updated:
10/09/03