The PC's in GBAD 311 and 416 have the HyperChem Windows program setup. The following directions are intended to get you started-for advanced applications consult the Help menus and the manual in GBAD 311. The topics covered in this handout are: construction of a molecular structure, geometric optimization for the structure, generation of the infrared spectrum, and animation of the virbrational modes. (The last two parts are only for demonstration.) It is easy to learn how to do things by just trying something. There is on-line help on the (surprise!) Help menu.
Sketch a Molecular Skeleton
1. Double left-click on the drawing tool (leftmost choice on the tool-bar).
2. Go to the main drawing screen (normally the black area) and build the carbon molecular skeleton by pointing to a spot and L(eft)-clicking. To erase an atom, point to it and R(ight)-click. To draw a bond to the next atom, L-click-and-drag.
3. Change the default element if needed ("Build," "Default Element"), to finish the rough molecular skeleton. Current versions also have convenient "Build with Carbon" or "Build with Oxygen" functions--to the right on the top icon bar.
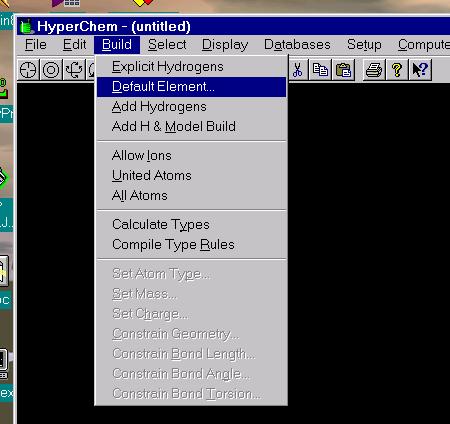
4. Close Element Table.
5. Make bonds by L-clicking and dragging between desired atoms. Click twice for double bond, etc. L-click three times on a ring bond to make the ring aromatic (dotted line).
6. Select the x-y rotation tool from the tool bar. (This gets you out of the drawing mode). You may click and drag to rotate the molecule. Other tools are Select, Rotate In Plane, Translate, and Zoom. The best way to learn to use them is to try!
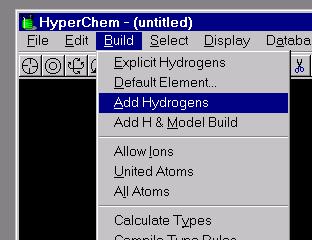
7. On the Top Menu Bar, select Build, Add Hydrogens (if hydrogens are needed). Next go to Build, Model Build to generate the standard 3-D structure of the molecule based on standard bond lengths and angles. The structure you have sketched out on the screen will be automatically updated. If you are happy with this structure, Save it to a File. HyperChem and other compatible modeling software (see manual) use binary files. The structure file has the extension .HIN.
You may find that the Model Build option gives you a bad configuration. A common example is that chair cyclohexanes wind up with axial instead of equatorial substituents. Stereochemistry may also get corrupted from what you want. To change configuration at an atom, select the Draw tool, place it over the atom, hold down the Shift key, and click on the atom.
9. Print is a choice under File. (Make sure the laser printer is On first!) Press the Print Screen key to copy whatever is on the screen to the Windows Clipboard. This screen copy can be pasted into the Windows Paintbrush program, and edited and printed, or saved from Paintbrush. Or, you can Copy the figure to the Windows Clipboard (under the Edit menu) and paste it into other Windows applications (WordPerfect, etc.)
Space Filling Rendering
At this point you can change the Display, Rendering to use Spheres (space filling) instead of the default Stick rendering. Note: use of Spheres (and even Disks) instead of Sticks requires a lot of video resources and may significantly slow things down if you rotate, show vibrations, etc. Also, there are several other choices, and a significant degree of customization is possible.
Calculations
Change the Display Rendering to Stick before starting any calculation (the program runs faster).
10. Select File, Start a Log File (extension .LOG) to keep track of the various methods tried and parameters generated during calculations. This information will be useful to you later.
11. Select Setup, Semiempirical, PM3. (You may want to examine results from different methods after looking at the PM3 results.) Accept the default parameters (OK).
12. Select Compute, Geometric Optimization. Accept any default parameters. After the optimization is done, save the updated structure file (File, Save). The energy gradient for the molecule has been minimized by slight changes in the geometry. If you want to refine the optimization, or change the criteria, consult the manuals.
13. All of this information from the calculation is located in the Log File, which you can open with any word processor (e.g., Notepad). Look for the following:
ENERGIES AND GRADIENT Total Energy = -40406.9247740 (kcal/mol) Total Energy = -64.391247746 (a.u.) Binding Energy = -2320.6224680 (kcal/mol) Isolated Atomic Energy = -38086.3023060 (kcal/mol) Electronic Energy = -217587.6753088 (kcal/mol) Core-Core Interaction = 177180.7505348 (kcal/mol) Heat of Formation = -104.9564680 (kcal/mol) Gradient = 9.2888496 (kcal/mol/Ang)
The calculated heat of formation for the molecule in a vacuum is the next
to the last line.
A quick way of finding ΔHf is to select
"Script" and go to the \c:\hyper directory. Select a script called
deltah.scr, and a dialog with the heat of formation will pop up. However, the log
file is your permanent record of the calculation.
Optional: Calculate the IR Spectrum
14. If you want to calculate the IR spectrum for your molecule select Compute, Vibrations. For your molecules, this process should take about 5-10 minutes. The progress is reported on the Status bar on the bottom of the window.
15. When the vibrational analysis is 100% done (look at the dialog window at the bottom of the screen), save the .HIN file, and close the Log file. Animate the Molecule after generating the Vibrational Spectrum
16. Use the Translational Tool to drag the molecule off to one side of the screen. Select Compute, Vibrational Spectrum (NOT the same as Step 14!). Drag the Vibrational Spectrum box to the other side of the screen. Highlight a vibrational mode by pointing to it and L-clicking. The IR active modes are at the bottom of the spectrum window, and Raman plus IR modes are on the top. Select Animate vibrations, Apply.
Note: If the structure file and vibrational parameters were generated at an earlier
time, load the optimized structure file, and run the vibrational script file that you
generated with the IR.SCR macro (default name was chem_ir.scr). Occasionally there are
spurious error messages when running this macro. Disregard these! Continue on, do not
abort the .SCR routine! If the macro has run successfully, Vibrational Spectrum
will be active under Compute. View and animate as above.
Last updated: 12/17/2014
Comments to K. Gable