The Diels-Alder reaction is a thermal cycloaddition. In the core reaction, a diene reacts with an alkene (called the "dienophile")to produce a cyclohexene ring.
Location of substituents and their stereochemistry follow from several empirical rules that are backed up by theory. We'll look at this in stages to develop these rules and explore a bit of what they mean.
|
The first thing to look at is where the substituents start, and where they wind up. The examples below show a transition state and both the initial (boat-like) form of the cyclohexene and the final chair-like form of the product. Critically, only a single diastereomer forms as the major product: this is stereospecific, and highly stereoselective.
|
Diels-Alder1TS.pdb
Show the interacting orbitals
The transition state. Atoms are color-coded in terms of their relationships in the reactants.
|
Diels-Alder_prod_boat.pdb
The initial form or the product. Note the retention of the stereochemistry in the monoene (red, blue), and the positional orientation of the brown and green atoms from the diene. The white atoms wind up on the cyclohexene double bond.
|
Diels-Alder_prod_chair.pdb
The initial product unfolds to a normal chair-like cyclohexene.
|
Controls:
Black background
White background
Teal background
Spacefilling model
Wireframe
Ball & Stick
|
This is a general observation, and coupled with the kinetic behavior (second order: first order in each reactant, or rate = k[diene][dienophile]), we can conclude that this is a concerted, one-step reaction mechanism. Any intermediate (radical or carbocation) would allow sigma bond rotation that would result in other diastereomers.
|
The next observation is that the dienophile could add in two possible ways to produce different diastereomers. In one, the electron-withdrawing (usually) substituent is oriented toward the newly forming C=C bond--this is the endo stereochemistry. In the other, it points away, and is called the exo stereochemistry. These are most easily shown in the two transition states for reaction of 1,3-cyclopentadiene with propenal (acrolein):
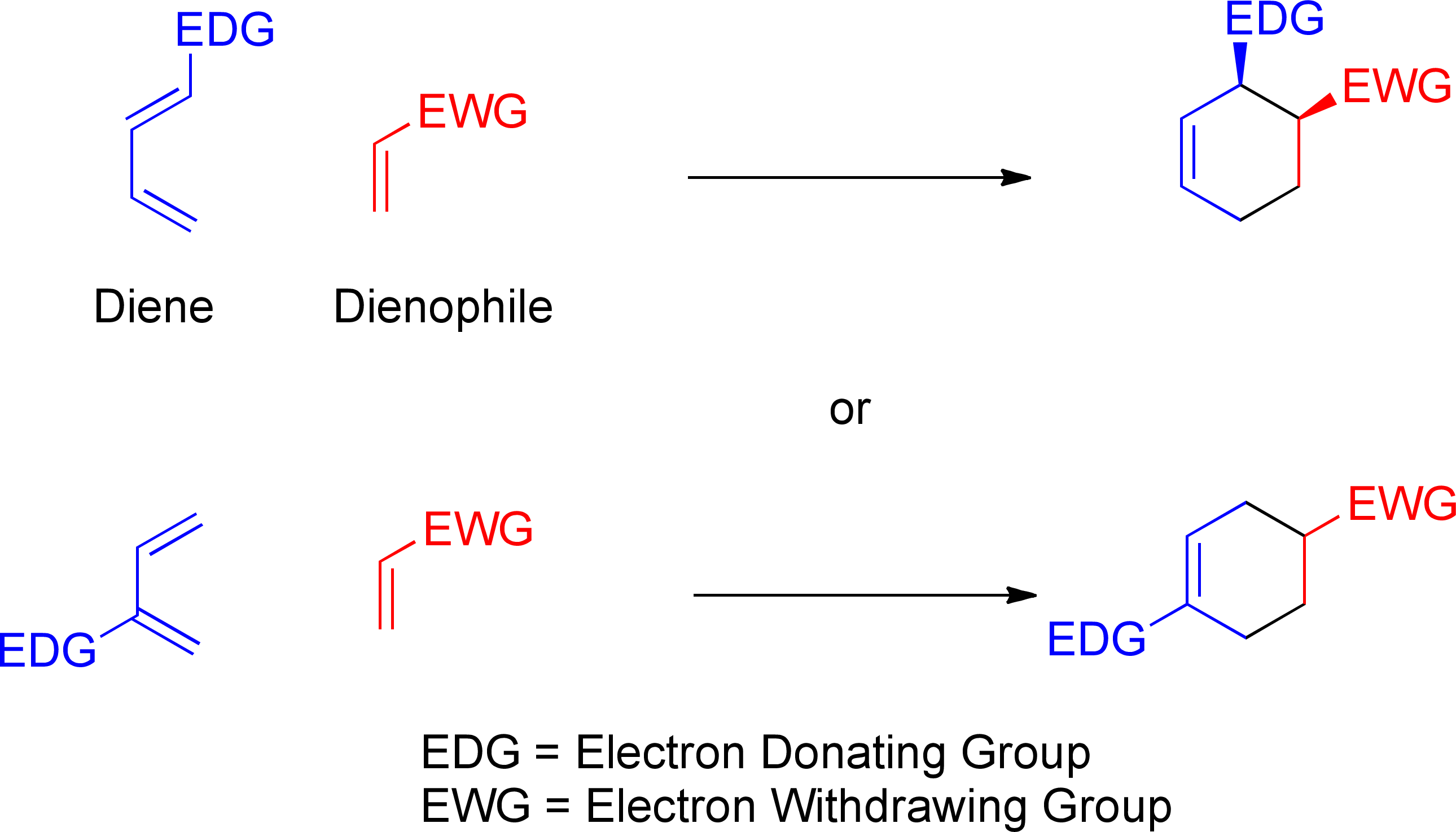
The final issue is the regiochemical orientation when there are two directing groups. Normally, the dienophile will have an electron-withdrawing group (lowers the LUMO and makes it a better electron acceptor); conversely, while unsubstituted dienes are fine (so long as they adopt the s-cis conformation about C2-C3), adding an electron donor (alkyl, OR, NR2) raises the HOMO and makes the diene a better electron donor. We have to worry about how best to orient these. A given pair (diene + dienophile) can have two possible regiochemical orientations:
- If the diene substituent is at position 1, the cyclohexene can have the two substituents either on adjacent carbons (formally the 3,4 pattern in the cyclohexene) or at nonadjacent positions (3,5). In a modestly inappropriate terminology in reference to aromatic compounds, this is a choice between "ortho" and "meta."
- If the diene substituent is at position 2, the cyclohexene can have the two substituents either at positions 1 and 5 ("meta") or at positions 1 and 4 ("para").
The empirical observation is that "ortho" and "para" are preferred over "meta," and this is borne out by calculated energies of the different transition states.
Last updated: 12/15/2019