MIPs As Chromatographic Stationary Phases for Molecular Recognition
Molecular imprint polymers recognize specific compounds and show promise as separation media, expecially for chiral molecules.
Oregon State University
One approach that seems promising is molecular imprinting, a technique for creating three-dimensional networks that have a "memory" of the shape and functional group positions of the template molecule. The resulting molecular imprint polymers (MIPs) can selectively recognize the template molecule used in the imprinting process, even in the presence of compounds with structure and functionality similar to those of the template. Molecular imprinting has become increasingly popular in recent years, and several excellent reviews have been published (4-9). MIPs have been applied as artificial antibodies (10), catalysts (11), sensors (9), drug assay tools (12), and chromatographic stationary phases (13). This Report focuses on the application of MIPs as separation media, especially as highly selective chiral stationary phases.
There is an increasing demand, especially in the agricultural chemicals and pharmaceutical industries, for better and more efficient means to prepare, purify, and analyze chiral compounds. This demand is driven by the sometimes markedly different biological activities of the enantiomers of a given compound and increasingly stringent regulations regarding optically active molecules. Despite dramatic improvements in asymmetric synthesis, chromatographic methods are still indispensable for analyzing and purifying chiral compounds. Commercial chiral stationary phases (CSPs) use immobilized chiral functionalities, ranging from small organic compounds to entire proteins. For separating a specific enantiomeric pair, several CSPs and mobile-phase conditions (specific to each CSP) may have to be evaluated to obtain satisfactory results; furthermore, the elution order of each enantiomer is often difficult to predict.
MIPs, in contrast, have predetermined selectivity and can be custom-made. When MIPs are used for chromatographic separations (Figure 1), the isomer used in the preparation of the MIP is always the one that is more strongly retained; therefore, the elution order is predictable. This is especially convenient for chiral separations, because further measurement would otherwise be necessary to determine the chirality of the eluted analytes.

Figure 1. Depiction of an HPLC column packed
with a MIP stationary phase used for chiral separations.
Many highly effective chiral separations can be achieved using conventional CSPs (14-17). MIP researchers hope to achieve one or more of several specific goals, including exquisite recognition and selectivity, production of low-cost media in bulk for preparative-scale separations, and the fabrication of a highly customizable sorbent. It is unlikely that MIPs will supplant the handful of CSPs that currently dominate the market and accommodate the majority of current chiral separation needs; nonetheless, MIPs offer unique advantages that will keep them at the focus of an expanding research field.
Basics
The concept of molecular imprinting was inspired by Pauling's antibody formation theory, in which an antigen is used as a template to aid in the rearrangement of antibody polypeptide chains so that the antibody has a configuration that complements the antigen molecule (18). Although Pauling's speculation was proven incorrect for antigen-antibody interactions, chemists found useful the idea of forming a three-dimensional structure around a template for preparing synthetic analogs of antibodies that can recognize target molecules with selectivities similar to their biological counterparts. The first experimental attempt at molecular imprinting was made in 1949 by imprinting a dye on silica gel (19).However, it was not until the early 1970s that successful imprints on synthetic organic polymers were achieved (4-8).
In molecular imprinting, monomers, such as methacrylic acid and styrene, are first assembled around the template molecule as a function of their complementary interactions. This arrangement of monomers is fixed on polymerization. The two general methods for establishing the prearrangement are covalent (4) and noncovalent (6) imprinting (Figure 2). During covalent imprinting, a monomer is linked to the template molecule via a labile covalent bond, such as boronic ester (4) or ketal (20).
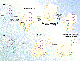
Figure 2. Simplified schematics of (a) covalent
and (b) noncovalent imprinting procedures.
The functionalized monomers are then copolymerized with an excess of crosslinker, such as divinylbenzene or ethylene glycol dimethacrylate, in the presence of a porogen (an inert solvent that encourages the formation of a network of pores in the resulting polymer). After polymerization, the polymer is dried, ground, and sieved; the "linker bond" is chemically cleaved, and the templates are freed from the polymer matrix, leaving cavities that are complementary to the template in shape and spatial configuration. An excess of crosslinker is ordinarily used to preserve and stabilize the specific cavities (4). Subsequent interactions between the template and the corresponding MIP can be in the form of either covalent (19) or noncovalent (21) interactions.
During noncovalent imprinting, weak intermolecular interactions are
used to self-assemble the monomer(s) around the template molecule. Typical
interactions include metal-ligand complexation; hydrogen bonding; and
ionic, ![]() -
-![]() , dipole, and hydrophobic
interactions. Because there is no covalent bonding between the template
and the monomers, the template is readily extracted after polymerization.
Care must be taken when choosing the monomers, crosslinkers, and porogens
in order to achieve good imprinting. The most commonly used monomers are
methacrylates, such as methacrylic acid, and heteroaromatic monomers, such
as vinylpyridine (6). Nonpolar aprotic solvents are generally used,
although attempts have been made to achieve noncovalent imprinting in an
aqueous environment (22).
, dipole, and hydrophobic
interactions. Because there is no covalent bonding between the template
and the monomers, the template is readily extracted after polymerization.
Care must be taken when choosing the monomers, crosslinkers, and porogens
in order to achieve good imprinting. The most commonly used monomers are
methacrylates, such as methacrylic acid, and heteroaromatic monomers, such
as vinylpyridine (6). Nonpolar aprotic solvents are generally used,
although attempts have been made to achieve noncovalent imprinting in an
aqueous environment (22).
The covalent imprinting approach is claimed to yield more uniform imprinted sites than the noncovalent approach because the monomer and template are held together by a chemical bond during polymerization. However, more quantitative determinations of uniformity are lacking. When the subsequent separation also relies on covalent interactions, the slow kinetics of such an interaction lead to extremely broad peaks and degrade resolution (4). The limited selection of reversible chemical reactions that can be used for covalent molecular imprinting also restricts the applicability of this method.
Noncovalent imprinting is more flexible and simpler to implement than covalent imprinting; thus, it has become the more popular method for synthesizing MIPs. Functional groups on both the monomer and the template are generally required to provide favorable interactions, and usually more than one interaction site on the template is required to achieve template-specific recognition. Good imprinting is usually accomplished via relatively strong noncovalent interactions; however, such strong interactions also cause extra broadening of the more retained peak when the finished MIP is used as a stationary phase.
In addition, because the assembly of monomeric species around the template is not strictly defined, the resulting binding sites have various affinities toward the template, similar to polyclonal antibodies. A fraction of the imprinted sites may provide the most favorable interactions with the template molecule and exhibit the highest affinity, whereas other imprinted sites may interact with the template in a less favorable or even a nonspecific manner. When such MIPs are used for chromatographic separation, the difference in the binding strengths of various imprinted sites gives rise to a nonlinear adsorption isotherm, which, in turn, results in extreme tailing of the more retained peak (23). These peak-broadening and tailing effects can be attenuated to an extent by careful optimization of the synthetic and chromatographic conditions. The mechanism of solute retention on imprinted polymers is, at best, quite poorly understood, although there is compelling evidence to support a cation-exchange mechanism in certain MIPs (23).
Making MIP stationary phases
MIPs can be prepared in several formats for use in chromatography. The
conventional approach is to synthesize the MIP in bulk, grind the
resulting polymer block into particles, and sieve the particles into the
desired size ranges. Particles of <25 ![]() m are generally used in
chromatographic studies. Such ground and sieved particles have been packed
into conventional HPLC columns (13, 22), immobilized on TLC
plates (24), and entrapped (25, 26) in capillary
columns using acrylamide gels or silicate matrices.
m are generally used in
chromatographic studies. Such ground and sieved particles have been packed
into conventional HPLC columns (13, 22), immobilized on TLC
plates (24), and entrapped (25, 26) in capillary
columns using acrylamide gels or silicate matrices.
Bulk polymerization is simple, and the optimization of imprinting conditions is relatively straightforward. However, the grinding and sieving steps are tedious, and only a portion of the polymer is recovered as useful packing material. The ground particles are polydisperse both in shape and size, which limits efficiency and resolution. Nevertheless, because of the inherent high selectivity of MIPs, complete enantioseparations have been achieved.
Efforts have been made to improve the shape and particle size distribution of MIP packing materials to attain better chromatographic performance (6). A layer of MIP has been grafted onto derivatized wide-pore silica particles and packed into HPLC columns (27). The small pores of the supporting silica particles may have been occluded by the MIP layer, thereby reducing the surface area available for interaction. This surface-imprinting approach may work better for imprinting bulky molecules such as proteins, which would not ordinarily sample the pore space to a significant degree anyway (21).
Polystyrene particles have also been used as shape templates for synthesizing MIPs via a two-stage swelling seed-polymerization technique (28, 29). Particles obtained using this technique are comparatively monodisperse in size and shape and well suited for chromatographic applications. However, the requirement for aqueous emulsions could interfere with the imprinting process and the selectivity of these particles is still not completely satisfactory. To avoid such interferences, suspension polymerization in perfluorocarbon solvents has been studied (30). Polymer beads produced using this method showed excellent chromatographic performance and good selectivity even at high flow rates. Unfortunately, the specialized perfluorocarbon solvent and fluorinated surfactant impose limits on the applicability and practicality of this method.
Another approach for preparing MIP stationary phases is to synthesize
the polymer in situ, thereby eliminating the need for packing the columns.
Matsui et al. made continuous-rod MIPs by heating 5 cm x 7.6 mm. i.d.
stainless steel columns filled with appropriate solutions at 70 ![]() C (31). Although columns
exhibiting reasonable selectivity were obtained in a single step, the poor
efficiencies resulted in low-resolution separations.
C (31). Although columns
exhibiting reasonable selectivity were obtained in a single step, the poor
efficiencies resulted in low-resolution separations.
Sellergren used a variant of the dispersion polymerization technique to prepare porous MIPs in 15 cm x 3 mm i.d. glass tubes (32). This approach gave rise to aggregates of micron-sized globular particles in the column. A mixture of isopropanol and water was used as the solvent for molecular imprinting. Chromatographic evaluation of this column showed a corrected selectivity of 6.8 between the template pentamidine (PAM, a drug used to treat AIDS-related disorders) and the reference compound benzamidine (BAM) at a flow rate of 0.3 mL/min (30). A corrected selectivity is determined by evaluating a nonimprinted polymer produced in a manner otherwise identical to the MIP under identical separation conditions. The MIP-based selectivity is then normalized to that of the nonimprinted polymer. An appreciable decrease in flow resistance was also observed for such columns; a flow rate of 5 mL/min was achieved at <1000 psi.
The dispersion polymerization method was also successfully applied to
the preparation of stationary phases in fused-silica capillaries (25 cm x
100 ![]() m i.d.) (33).
Electroosmotic flow was used to test these capillary columns. Complete
resolution of PAM and BAM was achieved, and significantly higher
efficiency for PAM was observed (N = 115,000 plates/m)
(32).
m i.d.) (33).
Electroosmotic flow was used to test these capillary columns. Complete
resolution of PAM and BAM was achieved, and significantly higher
efficiency for PAM was observed (N = 115,000 plates/m)
(32).
Porous monoliths of MIPs have also been prepared in situ in 35 cm x 75
![]() m i.d. fused-silica
capillaries using the nonpolar solvent toluene (34). The MIP
stationary phases were synthesized at low temperature (-20
m i.d. fused-silica
capillaries using the nonpolar solvent toluene (34). The MIP
stationary phases were synthesized at low temperature (-20 ![]() C) with UV initiation in an effort
to improve imprinting. This polymerization method necessitated the use of
capillaries with UV-transparent outer coatings, which tend to be more
expensive than the conventional polyimide-coated capillaries. The porous
nature of the monolithic polymer allowed easy hydrodynamic pumping during
the column preparation and conditioning steps. When these columns were
tested for use in capillary electrochromatography (CEC), complete
separation of a racemic mixture of the
C) with UV initiation in an effort
to improve imprinting. This polymerization method necessitated the use of
capillaries with UV-transparent outer coatings, which tend to be more
expensive than the conventional polyimide-coated capillaries. The porous
nature of the monolithic polymer allowed easy hydrodynamic pumping during
the column preparation and conditioning steps. When these columns were
tested for use in capillary electrochromatography (CEC), complete
separation of a racemic mixture of the ![]() -adrenergic
antagonist propranolol was accomplished in 2 min (34).
-adrenergic
antagonist propranolol was accomplished in 2 min (34).
MIP capillary columns can also be prepared as thin films for
open-tubular LC (35). This was achieved by in situ thermal
polymerization inside 25-![]() m
i.d. fused-silica capillaries. The low flow resistance of these columns
enabled separations at very low pressure (<1 bar/m of column length).
Both open-tubular LC and open-tubular CEC chiral separations of
dansyl-D L-phenylalanine were
demonstrated using the same commercial instrument.
m
i.d. fused-silica capillaries. The low flow resistance of these columns
enabled separations at very low pressure (<1 bar/m of column length).
Both open-tubular LC and open-tubular CEC chiral separations of
dansyl-D L-phenylalanine were
demonstrated using the same commercial instrument.
Chromatographic behavior
Although an extensive review of previous work done with MIP sorbents is beyond the scope of this Report, a brief foray into the growing literature will be helpful to gain an appreciation for the range over which this methodology may be used and for examining the strengths and weaknesses of MIPs.
Sample chromatograms obtained on columns packed with ground and sieved
MIP particles (<25 ![]() m) are
shown in Figure 3 (13). This particular MIP stationary phase was
prepared using typical reagents--methacrylic acid (MAA) was the functional
monomer, 2,2-bis(hydroxymethyl)butanol trimethacrylate (TRIM) was the
crosslinker, chloroform was the solvent, and (Z)-L-Ala-L-Ala-OMe was the template.
Under isocratic elution conditions and despite a selectivity of 1.92,
(Z)-D-Ala-D-Ala-OMe and
(Z)-L-Ala-L-Ala-OMe were
not completely resolved (Figure 3a) because of the broad and tailing
nature of the more retained peak (13). Excessive peak broadening
and tailing--a common failing of MIPs--were attributed to the polyclonal
nature of the MIP stationary phase, and gradient elution was used to
improve the separation (Figure 3b; apparent selectivity
m) are
shown in Figure 3 (13). This particular MIP stationary phase was
prepared using typical reagents--methacrylic acid (MAA) was the functional
monomer, 2,2-bis(hydroxymethyl)butanol trimethacrylate (TRIM) was the
crosslinker, chloroform was the solvent, and (Z)-L-Ala-L-Ala-OMe was the template.
Under isocratic elution conditions and despite a selectivity of 1.92,
(Z)-D-Ala-D-Ala-OMe and
(Z)-L-Ala-L-Ala-OMe were
not completely resolved (Figure 3a) because of the broad and tailing
nature of the more retained peak (13). Excessive peak broadening
and tailing--a common failing of MIPs--were attributed to the polyclonal
nature of the MIP stationary phase, and gradient elution was used to
improve the separation (Figure 3b; apparent selectivity ![]() = 3.19, resolution
RS = 4.50) (13). However, the apparent separation
efficiencies were still quite low (ND = 1154 and
NL = 3120 plates per meter), in part because of the
irregular shape and size dispersion of the MIP particles (13).
= 3.19, resolution
RS = 4.50) (13). However, the apparent separation
efficiencies were still quite low (ND = 1154 and
NL = 3120 plates per meter), in part because of the
irregular shape and size dispersion of the MIP particles (13).
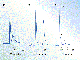
Figure 3. Chromatographic separations of
dipeptides on a column packed with ground and sieved polymer particles
(<25 ![]() m) imprinted with
(Z)-L-Ala-L-Ala-OMe.
m) imprinted with
(Z)-L-Ala-L-Ala-OMe.
The highly selective nature of this MIP stationary phase was also demonstrated by the separation of (Z)-D L-Ala-D L-Ala-OMe (Figure 3c) (13). As expected, the imprinted isomer, (Z)-L-Ala-L-Ala-OMe, was the most retained. An interesting secondary selectivity was also observed with this MIP stationary phase. It appeared that the MIP was capable of distinguishing the chirality of the first amino acid from the amino-terminus (separated (Z)-L-Ala- from (Z)-D-Ala-). In addition, when the first amino acid from the amino-terminus for an analyte was the same as the template ((Z)-L-Ala-), it could recognize the difference at the second amino acid (separated (Z)-L-Ala-D-Ala from (Z)-L-Ala-L-Ala)--an impressive testament to the recognition capabilities of imprinted polymers.
In addition, all of the dipeptides that had the same amino acid at the amino-terminus as the template were retained more than the other optical isomers. However, when the first amino acid from the amino-terminus was different from the template, the MIP was unable to distingish any difference at the second amino acid ((Z)-D-Ala-D-Ala coeluted with (Z)-D-Ala-L-Ala). It was concluded that the recognition of the amino-terminus of these molecules contributed more to the retention than the recognition of the carboxy-terminus (13). This conclusion is well founded, given that MAA, the functional monomer, will preferentially interact with the amino-terminus of the peptides under study. This observation also lends credence to the postulated cation-exchange mechanism of retention on many imprinted polymers.
Most MIPs prepared via the noncovalent imprinting method are synthesized and evaluated in nonpolar solvents. It has been shown that hydrogen bonding plays a significant role in the recognition processes of such MIP systems (36), in addition to the shape recognition (37). In one study, a MIP stationary phase imprinted with the dipeptide N-Ac-Phe-Trp-OMe was prepared in chloroform and evaluated in solvents of differing polarity (35). The selectivity observed in a polar solvent (such as acetonitrile) was lower than in a less polar solvent (such as chloroform). Furthermore, the inclusion of an aqueous mobile-phase additive reduced the contribution of hydrogen bonding toward analyte retention (35). Other studies have shown that ion-exchange and hydrophobic interaction effects are more significant than hydrogen bonding in highly aqueous mobile phases (38). The effect of pH on the capacity factor and selectivity of MIP stationary phases in mobile phases containing aqueous buffers has been studied, resulting in the previously mentioned hypothetical ion-exchange retention mechanism (21).
Another pertinent example of a MIP stationary phase was imprinted with
L-phenylalanine anilide (L-PA)
and synthesized in benzene, with MAA as the monomer and ethylene glycol
dimethacrylate (EDMA) as the crosslinker (23). Ground and sieved
MIP particles of 25- to 38-![]() m
nominal diameter were packed into a 10 cm x 5 mm i.d. column for
chromatographic evaluation. In a nonbuffered mobile phase, both D-PA and L-PA peaks were broad (Figure
4a). Although the selectivity was good (
m
nominal diameter were packed into a 10 cm x 5 mm i.d. column for
chromatographic evaluation. In a nonbuffered mobile phase, both D-PA and L-PA peaks were broad (Figure
4a). Although the selectivity was good (![]() = 5.4), the poor efficiency
(reduced plate heights of hD = 118,
hL = 397) limited the separation to
a resolution of only 1.1. (Reduced plate height is defined as the ratio of
plate height H to particle diameter dp.)
= 5.4), the poor efficiency
(reduced plate heights of hD = 118,
hL = 397) limited the separation to
a resolution of only 1.1. (Reduced plate height is defined as the ratio of
plate height H to particle diameter dp.)

Figure 4. Effects of mobile-phase pH and
temperature on elution profiles of D- and L-phenylalanine
anilide.
When a buffered mobile phase was used (Figure 4b), both efficiency
(hD = 75, hL = 167) and resolution (Rs = 1.3)
for D,L-PA improved, while the selectivity (![]() = 5.4) and the column
efficiency (h0 = 6, measured with a weakly retained
compound, acetone) remained the same as in Figure 4a. The MIP system was
quite sensitive to the pH of the aqueous component of the mobile phase, as
indicated in Figure 4c. The efficiency for the nonimprinted enantiomer was
slightly improved (hD = 70), but the selectivity (
= 5.4) and the column
efficiency (h0 = 6, measured with a weakly retained
compound, acetone) remained the same as in Figure 4a. The MIP system was
quite sensitive to the pH of the aqueous component of the mobile phase, as
indicated in Figure 4c. The efficiency for the nonimprinted enantiomer was
slightly improved (hD = 70), but the selectivity (![]() = 2.8) and the efficiency for
the imprinted enantiomer (hL = 1060) were considerably
poorer when the pH of the aqueous component of the eluent was reduced from
3.5 to 3.
= 2.8) and the efficiency for
the imprinted enantiomer (hL = 1060) were considerably
poorer when the pH of the aqueous component of the eluent was reduced from
3.5 to 3.
Additional improvement of column performance was achieved again by
adjusting the mobile-phase pH, using a heat-treated polymer as the
stationary phase and running the separation at an elevated temperature
(Figure 4d, hD = 9, hL = 18)
(23). The polymer was heat treated by subjecting the polymer to 120
![]() C for more than 12 h prior to
chromatographic evaluation. The heat treatment decreased the inhomogeneity
of binding affinity of the imprinted sites. Elevated column temperature
was later postulated to reduce the plate height contribution of slow
kinetics in analyte-stationary-phase interactions (38).
C for more than 12 h prior to
chromatographic evaluation. The heat treatment decreased the inhomogeneity
of binding affinity of the imprinted sites. Elevated column temperature
was later postulated to reduce the plate height contribution of slow
kinetics in analyte-stationary-phase interactions (38).
When MIPs are used as HPLC sorbents in packed columns, effective separations are accomplished mainly because of the inherent high selectivity of such stationary phases. However, column efficiencies are generally poorer than what is achievable on commercial chiral stationary phases. The separation efficiency of MIP systems can be improved by preparing the stationary phases in situ in the columns and using electroosmotic flow (33-35)--in other words, by operating in CEC mode. Unlike pressure-driven flow in HPLC, which has a parabolic flow profile, the electroosmotic flow in CEC has a plug-like profile, which can minimize the plate height contribution of packing and flow-velocity variations in the column (39). The nonlinear adsorption isotherm, however, will persist in limiting the attainable performance of these columns.
To date, the most rapid chiral separation achieved on a MIP stationary
phase was realized in the CEC mode (Figure 5) (34). A porous MIP
monolith imprinted with propranolol was prepared by photopolymerization in
a 75-![]() m i.d. fused-silica
capillary, using MAA as the monomer, TRIM as the crosslinker, and toluene
as the solvent. Racemic propranolol was separated in 2 min with a
resolution of 1.26. Under the same conditions, a neutral flow marker
(mesityl oxide) eluted at 3.3 min. The analytes were transported through
the column by both electrophoresis and electroosmosis.
m i.d. fused-silica
capillary, using MAA as the monomer, TRIM as the crosslinker, and toluene
as the solvent. Racemic propranolol was separated in 2 min with a
resolution of 1.26. Under the same conditions, a neutral flow marker
(mesityl oxide) eluted at 3.3 min. The analytes were transported through
the column by both electrophoresis and electroosmosis.

Figure 5. CEC separation of propranolol on
monolithic MIP stationary phase imprinted with (R)-propranolol.
Because the analytes carry positive charges at pH 3, they were eluted ahead of the neutral flow marker. However, both enantiomers should have the same electrophoretic mobility, which suggests that, in this instance, electrophoresis did not contribute to separative transport. The chiral separation resulted solely from chromatographic interactions between the analytes and the MIP stationary phase (34).
Thus far, the highest separation efficiency on a MIP stationary phase
was obtained using an open-tubular column (Figure 6) (35). A thin
layer of MIP stationary phase was prepared inside a 25-![]() m i.d. fused-silica capillary
using in situ thermal polymerization (40). The template molecule
was dansyl-L-phenylalanine, the functional monomers
were MAA and 2-vinyl pyridine, the crosslinker was EDMA, and the solvent
was a mixture of toluene and acetonitrile (35). Compared with
packed columns, higher efficiencies were expected in open-tubular columns
because of their high permeability and the absence of many of the
dispersive contributions typical of packed beds (41). When the
column was tested in the open-tubular LC mode with acetonitrile as the
mobile phase, the efficiencies for the nonimprinted and imprinted
enantiomers were 80,000 and 16,000 plates per meter, respectively
(35). A pressure of only 0.35 bar was required to establish bulk
flow because of the low flow resistance of the column.
m i.d. fused-silica capillary
using in situ thermal polymerization (40). The template molecule
was dansyl-L-phenylalanine, the functional monomers
were MAA and 2-vinyl pyridine, the crosslinker was EDMA, and the solvent
was a mixture of toluene and acetonitrile (35). Compared with
packed columns, higher efficiencies were expected in open-tubular columns
because of their high permeability and the absence of many of the
dispersive contributions typical of packed beds (41). When the
column was tested in the open-tubular LC mode with acetonitrile as the
mobile phase, the efficiencies for the nonimprinted and imprinted
enantiomers were 80,000 and 16,000 plates per meter, respectively
(35). A pressure of only 0.35 bar was required to establish bulk
flow because of the low flow resistance of the column.
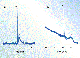
Figure 6. Open-tubular chiral separations of
1-mg/mL dansyl-DL-phenylalanine in acetonitrile on MIP stationary phases
imprinted with dansyl-L-phenyalanine.
When the column was tested in the open-tubular CEC mode, a different mobile phase containing an aqueous buffer was necessary to obtain a workable electroosmotic flow velocity. The relatively long analysis time was due to the column length (effectively 85 cm) and weak electroosmosis arising from the modest charge density at the polymer surface. The efficiency for the nonimprinted enantiomer was 249,000 plates per meter, more than three times that obtained in open-tubular LC mode, and was reproducible (35). The efficiency for the imprinted enantiomer, however, was only half that noted for the open-tubular LC separation. The decrease in efficiency for the imprinted analyte was attributed to possible changes in the mechanism of retention because of the increased aqueous component in the mobile phase (35).
Applications
The most extensive studies on MIPs as separation media were conducted
using amino acids (24-26, 35) and their derivatives
(13, 22-24, 28-30, 34, 36, 38) as the
template. Other classes of compounds that have been used in
chromatographic studies include peptides (13); sterols (20);
rigid aromatic compounds (19, 27, 30);
pharmaceuticals such as pentamidine (31, 32), ![]() -blockers
(33), and naproxen (42); pesticides (37); and sugars
and sugar derivatives (4, 43). High selectivity is generally
observed in separations using MIPs, whereas further improvements in
efficiency and kinetics are desirable. The molecular recognition ability
of MlPs has also encouraged their use as artificial antibodies
(10), biosensors (9), and catalysts (11).
-blockers
(33), and naproxen (42); pesticides (37); and sugars
and sugar derivatives (4, 43). High selectivity is generally
observed in separations using MIPs, whereas further improvements in
efficiency and kinetics are desirable. The molecular recognition ability
of MlPs has also encouraged their use as artificial antibodies
(10), biosensors (9), and catalysts (11).
Pros and cons
The synthetic nature and high degree of crosslinking of MIPs provide for unique chemical, mechanical, and thermal long-term stability relative to their biological counterparts, with the limitation that swelling and shrinkage cycles must be kept to an absolute minimum. The materials used for preparing MIPs are readily available and inexpensive, with the possible exception of the template. Template costs may vary considerably from one compound of interest to another. However, in both the covalent and noncovalent imprinting approaches, templates can be recovered in good yield and reused, reducing the cost per batch of MIPs. Relative to the design and synthesis of conventional surface-immobilized chiral stationary phases, preparation of MIPs is simple and easily scaled up, especially when the noncovalent imprinting approach is used. The selectivity of the MIP is predetermined and can be customized for a given separation.
The unfavorable adsorption isotherm and slow mass transfer in the polymer matrix are among the major limitations for MIPs as separation media. MIPs also tend to swell when the solvent composition is altered, which often leads to irreversible deformation of the imprinted cavity and loss of selectivity. A better understanding of the imprinting process and the recognition mechanism will certainly lead to improved imprinting techniques and enhanced chromatographic performance.
In most cases, MIPs are prepared and evaluated in organic solvents. Investigation of MIP performance in aqueous environments is desirable if they are to compete favorably with or complement biological recognition elements. In addition, only a limited number of studies have been done on the imprinting of macromolecules such as proteins. Surface imprinting in this area seems to be promising (21). Exploring new applications, synthetic methods, and materials, including highly rigid inorganic media (44), for MIPs as separation media will certainly increase their impact on analytical chemistry and other areas of research.
References
(1) Cram, D. J. Angew. Chem. Int. Ed. Engl. 1988, 27, 1009.
(2) Lehn, J. M. Angew. Chem. Int. Ed. Engl. 1988, 27, 89.
(3) Pedersen, C. J. Angew. Chem. Int. Ed. Engl. 1988, 27, 1021.
(4) Wulff, G. Angew. Chem. Int. Ed. Engl. 1995, 34, 1812.
(5) Steinke, J.; Sherrington, D. C.; Dunkin, I. R. Adv. Polym. Sci. 1995, 123, 81.
(6) Mayes, A. G.; Mosbach, K. Trends Anal. Chem. 1997, 16, 321.
(7) Vidyasankar, A. Current Opinion in Biotechnology 1997, 16, 310.
(8) Sellergren, B. Trends Anal. Chem. 1997, 16, 310.
(9) Kriz, D.; Ramstrom, O.; Mosbach, K. Anal. Chem. 1997, 69, 345 A.
(10) Ansell, R. J.; Ramstrom, A. O.; Mosbach, K. Clin. Chem. 1996, 42, 1506.
(11) Beach, H. V.; Shea, K. J. J. Am. Chem. Soc. 1994, 116, 379.
(12) Vlatakis, G.; Andersson, L. I.; Muller, R.; Mosbach, K. Nature 1993, 361, 645.
(13) Kempe, M. Anal. Chem. 1996, 68, 1948.
(14) Ekborg-Ott, K. H.; Kullman, J. P.; Wang, X.; Gahm, K.; He, L.; Armstrong, D. Chirality 1998, 10, 627.
(15) Liu, J.; Stewart, J. T. Anal. Lett. 1987, 30, 1997.
(16) Verleysen, K; Sandra, P. J. High Resolut. Chromatogr. 1999, 22, 33.
(17) Majors, R. E. LC-GC 1997, 15, 412.
(18) Pauling, L. J. Am. Chem. Soc. 1940, 62, 2643.
(19) Dickey, F. H. Proc. Natl. Acad. Sci. U.S. 1949, 35, 277.
(20) Shea, K. J.; Sasaki, D. Y. J. Am. Chem. Soc. 1991, 113, 4109.
(21) Whitcombe, M. J.; Rodriguez, M. E.; Villar, P.; Vulfson, E. N. J. Am. Chem. Soc. 1995, 117, 7105.
(22) Burow, M.; Minoura, N. Biochem. Biophys. Res. Commun. 1996, 227, 419.
(23) Sellergren, B.; Shea, K. J. J. Chromatogr., A 1993, 654, 17.
(24) Kriz, D.; Kriz, C. B.; Andersson, L. I.; Mosbach, K. Anal. Chem. 1994, 66, 2636.
(25) Lin, J. M.; Nakagama, T.; Uchiyama, K.; Hobo, T. Chromatographia 1996, 43, 585.
(26) Chirica, G.; Remcho, V. T. Electrophoresis 1999, 20, 50.
(27) Vidyasankar, S.; Ru, M.; Arnold, F. H. J. Chromatogr., A 1997, 775, 51.
(28) Hosoya, K., et al. J. Chromatogr., A 1996, 728, 139.
(29) Tan, Z. J. Ph.D. Dissertation, West Virginia University, Morgantown, WV, 1998.
(30) Mayes, A. G.; Mosbach, K. Anal. Chem. 1996, 68, 3769.
(31) Matsui, J., et al. Anal. Chem. 1993, 65, 2223.
(32) Sellergren, B. J. Chromatogr., A 1994, 673, 133.
(33) Nilsson, K.; Lindell, J.; Norrolow, O.; Sellergren, B. J. Chromatogr., A 1994, 680, 57.
(34) Schweitz, L.; Andersson, L. I; Nilsson, S. Anal. Chem. 1997, 69, 1179.
(35) Tan, Z. J.; Remcho, V. T. Electrophoresis 1998, 19, 2055.
(36) Nicholls, I. A.; Ramstrom, O.; Mosbach, K. J. Chromatogr., A 1995, 691, 349.
(37) Kempe, M.; Mosbach, K. Int. J. Pep. Protein Res. 1994, 44, 603.
(38) Dauwe, C.; Sellergren, B. J. Chromatogr., A 1996, 753, 191.
(39) Colon, L. A.; Guo, Y.; Fermier, A. Anal. Chem. 1997, 69, 461 A.
(40) Tan, Z. J.; Remcho, V. T. Anal. Chem. 1997, 69, 581.
(41) Scott, R. W. J. Chromatogr., A 1990, 517, 297.
(42) Kempe, M.; Mosbach, K. J. Chromatogr., A 1994, 664, 276.
(43) Wulff, G.; Schauhoff, S. J. Org. Chem. 1991, 56, 395.
(44) Pinel, C.; Loisil, P.; Gallezot, P. Adv. Mater. 1997, 9, 582.
Vincent T. Remcho is associate professor at Oregon State University. His research interests include microscale separations, molecular recognition processes, specialized chromatographic media, and flow dynamics in separations systems. Z. Jessica Tan is a postdoctoral fellow at the University of Illinois at Urbana-Champaign. Her current research efforts encompass separations and NMR. Address correspondence to Remcho at Department of Chemistry, Oregon State University, Corvallis, OR 97331-4003 (remchov@chem.orst.edu).