A large component of Dr. Gable's research strives to understand organotransition metal chemistry in a manner that will lead to new catalytic transformations of organic molecules. This involves making organic compounds as substrates, and inorganic and organometallic compounds as reactants. The area that dominates this work is fragmentation of oxametallacycles as mechanistic probes for oxidation catalysts.
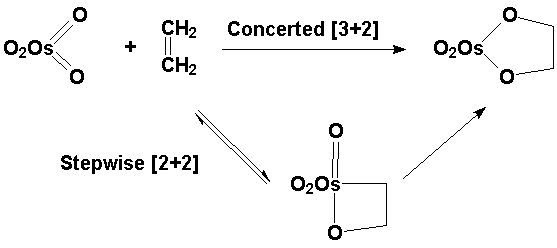
The elementary processes by which an alkene interacts with a metal oxide are currently poorly understood. One area of controversy is whether a metallaoxetane is involved in reactions such as bishydroxylation by OsO4:
Our approach has been to look at a model system. Several rhenium diolate complexes extrude alkenes, a process that is the microscopic reverse of osmylation. The goal has been to characterize the transition state for the extrusion as a means of distinguishing the concerted, pericyclic pathway from a stepwise process involving a metallaoxetane:
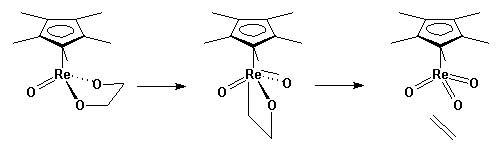
Experimentally, we measure reaction rates as a function of structural changes in the molecule. We also use isotopic labeling, kinetic isotope effects, and other tools of physical organic chemistry to characterize the behavior of the system. Several lines of evidence lead to the conclusion that this process is stepwise. The most important of these is the effect of strain on the reaction energetics. Strain energy in the double bond has no effect on the energetics of alkene extrusion, but accelerates the oxidation so that Cp*ReO3 will oxidize norbornadiene and trans-cyclooctene. This implies there is no rehybridization of carbon to sp2, and suggests asynchronous C-O cleavage.
Recent theoretical work, though, has cast doubt on whether the stepwise mechanism is viable. A rigorous test involves comparing isotope effects for different reaction sites in an asymmetric substrate:
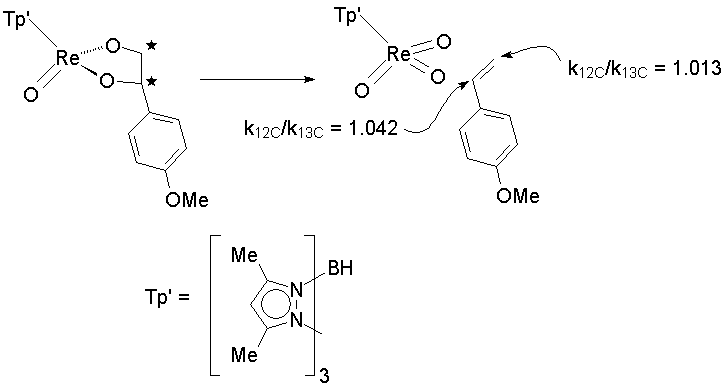
The observation of KIEs of greater than one at both positions demands that both C-O bonds break, to some extent, in the transition state. However, the inequality implies a substantial asymmetry in the degree of bond breaking. This is supported by a theoretical model of the transition state structure (LACVP*+/B3LYP):
A related project involves the O-atom transfer properties of LReO2 complexes. We showed that while the stoichiometric reaction of {Cp*ReO(μ-O)}2 with epoxides leads to clean O-atom transfer, a catalytic form of the reaction was quite inefficient, due to formation of a novel purple cluster compound. We believe this suggests the neutral {Cp*ReO(μ-O)}3 is also a component of the monomer-dimer equilibrium, and that trapping this trimer may be possible using other metal complexes. Under the conditions of the catalytic reaction, further O-atom transfer oxidizes this tetranuclear cluster to a green trinuclear cluster (isolated as a perrhenate).
On the left is our X-ray crystal structure of the tetranuclear cluster; on
the right is the eventual oxidized cluster (a dication), which had been previously
reported by the Herrmann group from Munich.
Tetranuclear cluster (+1 cation) |
Trinuclear cluster (+2 cation) |
Current work is focused on the ability of several LReO2 compounds to abstract oxygen from epoxides, sulfoxides, phosphine oxides and other O-atom donors. We are exploring issues such as the influence of the ligand on the structure and properties of the dioxo compound, the influence of water and other competing ligands on the O-atom transfer, and particularly in whether the mechanism of this atom transfer reaction can be related to that of the diolate cycloreversions. Preliminary work indicates that different epoxide-binding modes can direct the complex into different reaction pathways.
This effort involves measuring reaction rates for specific steps thought to be part of the catalytic cycle. Future work involves exploring related reactions with other heteroatoms, and implementation of chiral ligands to effect stereoselective atom transfer and kinetic resolution of racemic O-atom donors. Work is in progress to demonstrate the utility of catalytic diol deoxygenation for transforming biomass-derived carbohydrates into other useful organic compounds.
To learn more about our work in this area, check out the following recent publications. (Note that some of the electronic links may require paid subscriptions; for a free reprint please email me. )
Click here for a full listing of papers.
This work has been supported by the National Science Foundation.
Back to Kevin Gable's Home Page
Last updated by K. Gable 02/20/2014
Comments to K. Gable
Page made with