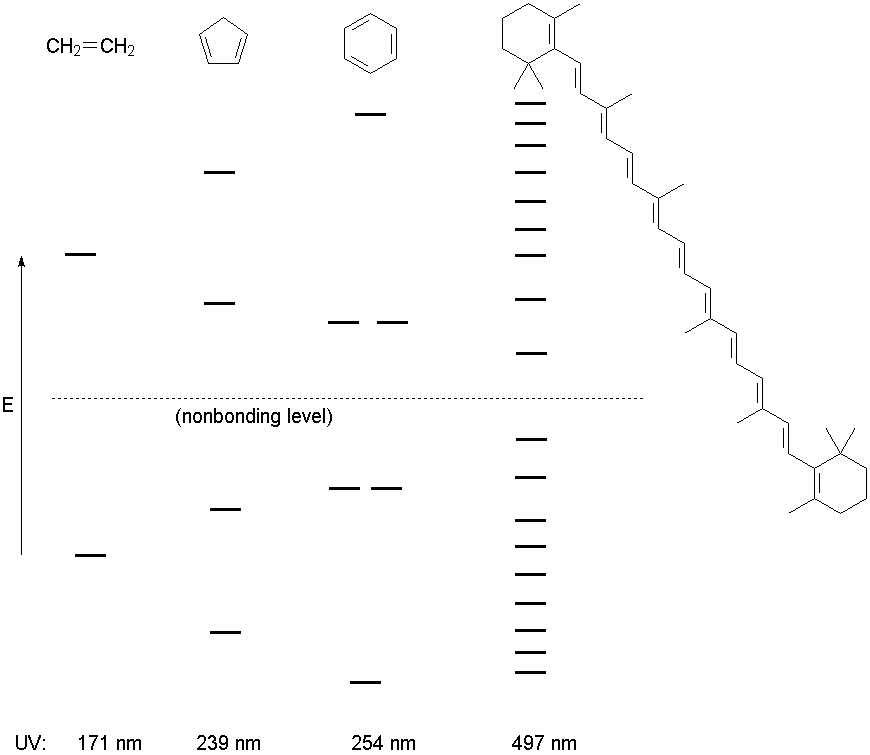|
|
CH336Conjugated SystemsApplication of MO Theory |
 |
|
|
||
|
| ||
|
| ||
|
| ||
|
|
|
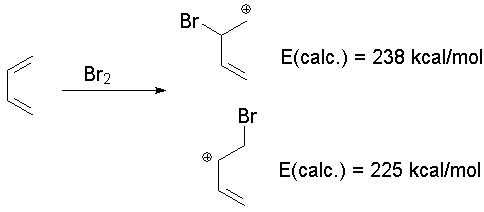
There are two major uses of the orbital pictures and energies generated from MO theory. One is to predict reactivity, and the other is to predict properties like UV spectra. Prediction of ReactivityElectrophiles will react with the most reactive electrons in the molecule, usually. The Highest Occupied Moleculat Orbital (HOMO) tells us where these electrons are. The Lowest Unoccupied Molecular Orbital (LUMO) tells us where any additional electrons (i.e., from a nucleophile) can go. We can also compare the energy of isomeric structures to predict reactivity. Let's look at bromination of butadiene. The HOMO of butadiene is not really helpful, since it shows density at both C1 and C2. But MO theory allows us to predict the relative energy of reacting Br+ at either position (a full calculation is necessary to get this information):
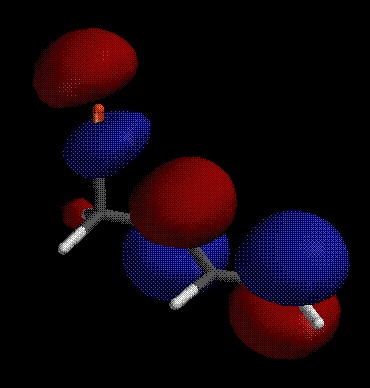
The calculations are for gas phase molecules, and do not consider solvation, so these cations both have very positive heats of formation. However, the calculations predict (accurately) that addition of the electrophile to the C1 is more favorable than addition to C2 by about 13 kcal/mol. We can also look at the LUMO of the preferred isomer to see where a nucleophile is likely to bond:
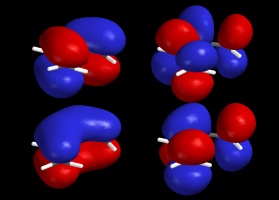
Because of the orbital density at carbons 2 and 4, we expect a nucleophile to add at either position to give a mixture of isomeric products. Recall that using resonance structures gives a very similar prediction, both as to the regiochemistry of electrophile addition and the possibile formation of isomeric products (1,2- and 1,4-addition). UV SpectroscopyMolecules absorb visible and ultraviolet radiation when the energy of the light matches the energy of an electronic transition in the molecule. In organic molecules, the important absorbances promote electrons from the HOMO or other high-lying molecular orbitals to the LUMO or other low-lying empty orbitals. In unsaturated organic molecules, the HOMO is usually a p bonding orbital, and the LUMO is an antibonding orbital involving the p system, designated p* because of its antibonding nature. Note that resonance theory does not give you many tools for predicting UV behavior; the model fundamentally treats electrons as localized bonding (or lone) pairs, and tends to ignore the possibility of empty orbitals to which electrons can be excited. Thus MO theory is critical to understanding the physical origin of UV/visible light absorbance, and particularly to the prediction of structural effects on the UV spectrum. The energy level diagram for several unsaturated systems is shown below.
|