1. Make sure your data files are in a directory on the computer on which you are working. For example, transfer them from the network drive to a USB drive.
2. Open NUTS by clicking on the Acorn icon on one of the PC's in GBAD311
3. Open your data file. The raw data, or Free Induction Decay (FID), should come up on
the screen. This is the interferogram.
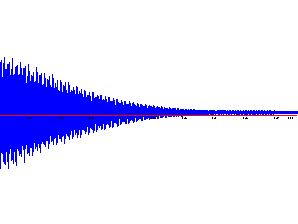
Raw data: When you open the FID, it looks like this:
|
|
This is an intensity-vs.-time plot. |
One minor adjustment to the data must be made. Type RD, and hit "OK" on the dialog box that pops up. This modification ensures that the data as produced by the spectrometer is converted to a mathematical form that NUTS is expecting.
4. Perform a Fourier Transform on the FID by going to "Processes"; "Transforms"; "Fourier Transform." In a second or two, the FID will be transformed from a time-based to a frequency-based spectrum.
Transformed data: Then, after you perform the Fourier Transform, you will normally see something like:
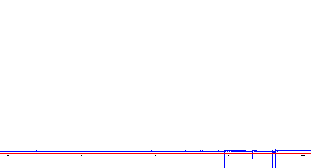 |
The peaks in the left spectrum are upside down! |
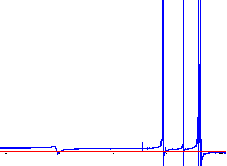 |
The right spectrum is more typical. |
The peaks in the typical display are uneven from side to side and dip below the baseline. Since the goal here is to be able to accurately integrate the peaks, the peaks first need to be phased to remove the asymmetry as much as possible. This is what Phasing is all about.
5. Normally some phasing of the spectrum will be necessary. This is one of the more difficult skills in working up NMR data. The following directions will lead you through the processes of phasing and then integration of the spectrum.
Phasing:
A. Go to Process:Phasing, and select Quick Phase. You will get a pretty good spectrum from this (left), but if you expand the Y-axis (scroll bar on the right), you will see that the peaks are not really phased properly (right spectrum below): (The curvature in the baseline is normal; you will take care of this after phasing.)
|
|
Typical result from Quick Phase. |
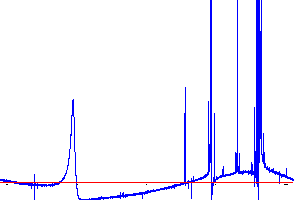 |
Expanded Y-axis reveals that there is still work to be done! |
B. Now, use Phase by mouse. This uses both the left and right mouse buttons. In sequence, click and drag the left button left or right to phase a peak on the left side of the spectrum. Go a little bit "too far". Try to make all peaks have the same shape. Then right click and drag to phase the right side of the spectrum. It is usually necessary to go back and forth several times to get a good phase:
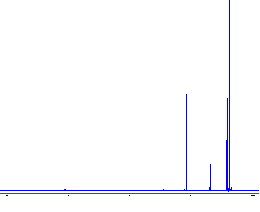 |
This spectrum is just right! |
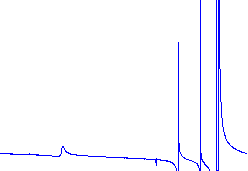 |
This spectrum needs a better phase adjustment. |
Important! Hit "Enter" (or you'll lose the phase correction!).
C. Additional Directions for NUTS that covers phasing two separate regions are given next. You can also consult the Help in NUTS.
1. Select Phase by Mouse (main menu-Process.) Use the Y-axis scroll bar to zoom the intensities around the most upfield peak. Phase this signal using the mouse. Phase by mouse works pretty good for the signals around 2 ppm.
2. Usually it not possible to phase the entire spectrum at once so use the Phase Expansion procedure given next. In it you will set two separate regions of the spectrum and phase them separately. For example you will want to use one of the middle peaks and the most downfield peak (OH) for this part.
Type ZO (use caps)
Type Cntl-F This selects the entire spectrum to be viewed (the line turns blue)
Use the left mouse button and select region 1 by left clicking and dragging over the signal around 5 ppm (the selected region will appear in red) To set this as region 1
Type 1
Use the left mouse button to click and drag over region 2 (use the OH region around 15 ppm)
Type 2
Type PE (use caps)
Right click to view and phase region 1 and Left click to view and phase region 2
Use the mouse to phase each peak separately, when you are satisfied with the phase, press Enter. If you don't do this, all your phasing work will be lost!
3. Go to View and select "Show all reals". This re-displays the entire spectrum. Inspect the phasing. Fix phase if needed on remaining signals using the simple phase by mouse, or if more serious phasing is needed, repeat PE procedure.
If you want more information on this procedure, see NUTS help files for phasing.
Integration of Peaks:
Baseline Correction: Accurate integration will require a good baseline. To
correct the slight baseline curvature normally present, go to "Processes";
"Fit Baseline". A series of vertical red stripes, corresponding to what the
program thinks is baseline, overlies the spectrum. If by chance a real peak lies in one of
the red areas, mouse click on it to remove it from consideration. Use as many sections as
possible; normally the initial selection is OK.
Type "c" to begin the calculation; when the calculation is finished type
"a" to apply the correction and return to normal mode. Your spectrum should look
like this:
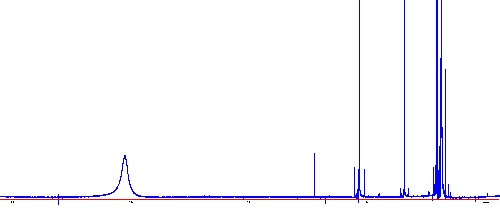
Establish a reference for the chemical shift axis (X-axis):
A. Set the solvent reference peak by placing the cursor on the solvent peak, hold the left-click as you type "V". A dialog box for inputting the chemical shift for the proteated solvent peak will appear. Enter the correct chemical shift value for the solvent peak that you have selected: (for additional values click here) This same solvent peak will be used to reference the integrations in the next part.
| CHCl3 | 7.26 ppm | C6D5H | 7.15 ppm | CD2HOD | 3.30 ppm |
| CD2HCN | 1.94 ppm | CD2HC(O)CD3 | 2.04 ppm | CD2HS(O)CD3 | 2.50 ppm |
B. Save the file using the Save As command. Next you will want to Integrate the signals.
Integration:
1. Type ZO then f (note cases, and do not press enter.) Set the integration limits in the prompt box that appears on the screen as follows:
use 2.5 ppm for the "high" limit and 1.0 ppm for the "low" limit.
2. Right click the mouse.
3. Type ID (caps) This displays the entire spectrum and an integral trace over the entire spectrum.
4. Use the left mouse button to set the range over each peak for integration, one peak at a time. To do this, Left click on the left side of the peak (line turns green), then Left click again and drag to the right side of the peak (line turns red), Left click to set. The integral over the peak will appear on the screen. Try to avoid selecting 13-C satellite peaks and spinning side bands (these are symmetrical around the main signal.)
5. Use the X-axis scroll bar to scroll to the next signal and integrate it. Repeat procedure until all the peaks have been integrated. Also, integrate the solvent reference peak.
6. When done integrating all the signals, return the mouse to the reference peak and left click and hold right on the top of the solvent peak hold down and press the "v" key and a prompt box will come up. In the prompt, set the value for the solvent peak integral to 1.00 . (very important, don’t double click or release until next step is done, or all work can be lost.) This establishes a reference area for all the other integral area values and all peaks will be labeled with relative values for the peak areas.
7. Press Enter. This accepts the integrals and exits ID.
8. Go to View - Show Integrals
9. Go to View - Show all Reals. Congratulations!! You should see a normal spectrum with signals integrated and labeled!
Saving and Printing:
When the spectrum is as you want it, save it and print under the "File" menu. To print from NUTS to the new 2200TN laser jet in GBAD 311(winter term 2002) make sure that the following choices are selected for printer setup within NUTS. You have to set these parameters each time you open NUTS, but they stay active during the session. First, select File-Printer Dialog, then
Select Finishing tab- select Print Quality-Best
Select Effects tab-Print Doc on (check box); select Letter; select Scale to Fit
Select Basics tab- pick Landscape
Note: the first time you print, some labels may be missing, send the job a second time and it should work fine.
Note that expansions may be obtained with the Zoom routine and copied and pasted using the appropriate Windows functions, or (better) printed separately. If at any point you mess up, you can always return to the full spectrum with "View"; "Show All Reals". Or you may reload the original data and re-transform. You can also copy Metafile to the Windows clipboard and copy into a wordprocessor.
If you want to practice this on your own computer, a demo version of NUTS is available (with some sample spectra) from Acorn NMR: http://www.acornnmr.com/. You will not be able to copy or print from this version, but it will give you some experience with using the program to manipulate data.
Acknowledgements:
Kevin Gable for figures and much of text.
Emile Firpo for detailing the procedure to use NUTS to phase and
integrate FID raw data from the Bruker 400DP.
Return to CH 362 home page